
I'm looking for information about
Can't find what you're looking for? You may need to login to see more documents
G
Guidance
Suggested parameters and sets of instructions outlining best practices and standards for accomplishing specific duties.
Suggested parameters and sets of instructions outlining best practices and standards for accomplishing specific duties.
This Copy Was Generated On: July 28, 2026
Adverse Event Reporting
IRBMED
| Approval Date:
June 11, 2018 8:00 am
Internal adverse events are events that involve subjects or others whom the U-M investigator is responsible for and/or events for which a UM IRB has direct oversight.
These guidelines apply only to IRBMED reporting. They do not apply to required reporting to other internal or external oversight agencies or departments (e.g., sponsor, FDA, UMHS Compliance Office, DSMB, or Privacy Offices).
For information about other types of required reporting, see the Adverse Events (AEs), Other Reportable Information and Occurrences (ORIOs), and Other Required Reporting page.
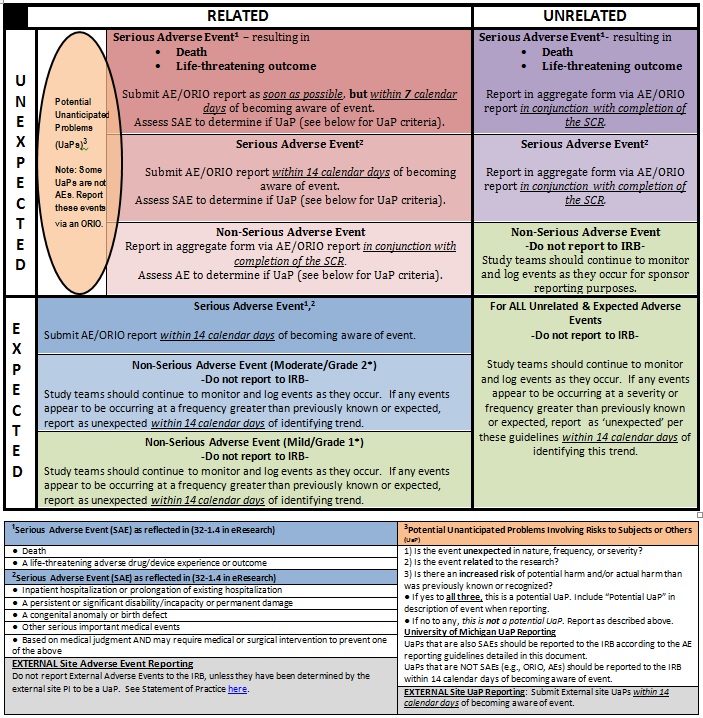
Reporting timetable for approved studies subject to annual Continuing Review (CR)
“Continuing Review” (CR) consists of annual submission by study team of a report on the study’s status and continued plans, for renewed review and approval by the IRB. See IRBMED FAQ heading “Ongoing Review.” If an approved study space shows an Expiration Date, the study requires CR.
This chart is for studies requiring annual Continuing Review that follow IRBMED standard AE reporting. It may be appropriate for some studies to consider a Study-Specific AE Reporting Plan. See the gray boxes below for information about External AE and UaP reporting.
Definitions
SCR: Scheduled Continuing Review; Expected: Has been addressed in one or more of following: Protocol, Investigator Brochure, Package Insert or equivalent, published literature, IRB application, grant application, Data Safety Monitoring Board/Data Safety Committee reports, other documentation, informed consent document (ICD) or characteristics of the study population. Note, per OHRP guidance, event may be “expected” per the natural progression of any underlying disease, disorder, or condition of the subject(s) experiencing the adverse event; Unexpected: Has not been addressed in one or more of the above examples; Related: See below; Unrelated: See Below
References
Defining Relationship to Study Drug/Device/Procedure
The IRB recognizes that it can be difficult to determine the relationship of event or events to a specific drug/device/procedure when there are several contributing factors. Events should be rated according to the following parameters:
Studies NOT requiring Continuing Review (CR)
Per 2018 Revised Common Rule, some ongoing studies no longer require annual Continuing Review (CR). The study workspace indicates “No Continuing Review Required for this application” when applicable. Studies not requiring CR should report only potential Unanticipated Problems (UaPs or UPIRSOs) that come to the attention of the study team. Routine AEs need not be reported. This also applies to studies that previously had an approved Study-specific AE reporting plan.
Unanticipated Problems meet all of the following criteria:
- Unexpected (in terms of nature, severity, or frequency);
- Related or possibly related to participation in the research; and
- Suggests that the research places subjects or others at a greater risk of harm than was previously known or recognized.
See the Unanticipated Problems Involving Risks to Subjects or Others page and U-M HRPP Operations Manual Part 12.II.B for more details.
Events Related
Definitely Related
- The event is a known effect of the drug, device, or procedure (e.g., listed in the protocol documents including IB, consent, publications)
- The event follows an obvious sequence of time, from the drug’s administration, device’s implantation or activation, or procedure, for which the event is directly attributed to the administration, implantation, activation, or procedure.
- The event ceases with discontinuation of the drug, device, or procedure (and reoccurs on restarting).
- The event includes data that was only collected for the study.
- The event included disturbing or upsetting questions that the subject was asked for the purpose of the
research.
Probably Related
- The event is lesser known or suspected effect of the drug, device, or procedure (listed in the protocol documents including IB, consent, publications, etc.)
- The event follows a reasonable sequence of time from the drug’s administration, device implantation, activation, or procedure, for which the event may be attributed to the administration, implantation, activation, or procedure.
- The event ceases or diminishes with discontinuation of the drug, removal/discontinued activation of the device, or procedure.
Possibly Related
- The event is a lesser known or possible effect of the drug, device, or procedure.
- The event occurred within a sequence of time from the drug’s administration, device implantation and/or activation, or procedure, for which the event may be attributed to the administration, implantation, activation, or procedure.
- The event could be explained by the characteristics of the population under study.
Events Not Related
Unlikely Related
- The event is NOT a previously known or suspected effect of the test drug, device, or procedure.
- The event does NOT follow a sequence of time# from drug administration, device implantation and/or activation, or procedure, for which the event could be attributed to the administration, implantation, activation, or procedure.
- The event can be readily explained by the characteristics of the population under study.
Unrelated
- The event is NOT known to be an effect of the test drug, device, or procedure.
- The event does NOT follow a sequence of time from drug administration, device implantation and/or activation, or procedure, for which the event could be attributed to the administration, implantation, activation, or procedure.
- The event can be readily and easily explained by the characteristics of the population under study.
- Subject never received study drug, study device, or underwent research study procedure.
Step-by-step instructions (below) for applying these guidelines appear below.
Other Reporting Tables
- Study-Specific Adverse Event (AE) Reporting
- Encouraged for minimal- to moderate-risk studies that are not under FDA oversight; many sponsored studies include an AE reporting plan.
- External/Non-UM AE Reporting
- Humanitarian Use Device (HUD) under a Humanitarian Device Exemption (HDE)
- Other Reportable Information and Occurrences (ORIO)
- Events that are not adverse events (no harm occurred) or reports to or from and oversight bodies
Reporting Procedures
For studies in eResearch (IRB web-based application system):
Open the approved study in eResearch. Click on AE/ORIO function tab in the left column (which looks like the screen shot shown to the here).
- Instructions for submitting an AE are available here.
- If the event necessitates an amendment to the protocol or informed consent document, you must submit an amendment after submitting the AE. If the AE obliges the investigator to inform previously enrolled subjects, include in the amendment the mechanism by which this will be done.
- For example, sending a letter to all previously enrolled subjects, upload the letter in the amendment submission.
Follow these steps if you are reporting a U-M Event
| Review Step 1 | The principal investigator should assess each adverse event by the following criteria. Note the answers, then refer to the timetable to determine the deadline by which the report should be submitted to the IRB. | |
| Step 2 | Do the IRB approved documents for this study include a detailed plan with a specific timetable or time schedule for AE reporting? (eResearch studies, see approved application section 32-1) | |
| YES: Use the Study Specific AE Reporting Timetable | NO: Continue below to Step 3 | |
| Step 3 | Is the AE to be reported addressed or described in one or more of the following?
|
|
| YES the AE to be reported is EXPECTED | NO the AE to be reported is UNEXPECTED | |
| Step 4 | Determine the association of the AE to be reported with the test article or test procedure (the interaction or intervention):
RELATEDNESS refers to the cause of the adverse event. Investigators should assess whether the event is attributable, in whole or in part, to the study intervention/agent. If the investigator judges the event as being PROBABLY related to a concurrent standard therapy, but POSSIBLY related to the investigational therapy, report the event as “possibly related.” Indicate on the reporting form the UM PI judgment regarding which part of the therapy the event should be attributed to. If the PI and sponsor disagree on the assessment, so note in the report. |
|
| Step 5 | Based on the event’s relatedness and expectedness, report it within the timeframe indicated on the Standard Adverse Event Reporting chart above. | |
Questions?
Contact us at [email protected] or 734-763-4768 / (Fax 734-763-1234)
2800 Plymouth Road, Ann Arbor, MI 48109-2800
A list of IRBMED staff is available at our website.
Edited By: [email protected]
Last Updated: February 23, 2026, 11:15 AM
See Also
G
Guidance
Suggested parameters and sets of instructions outlining best practices and standards for accomplishing specific duties.
Suggested parameters and sets of instructions outlining best practices and standards for accomplishing specific duties.
G
Guidance
Suggested parameters and sets of instructions outlining best practices and standards for accomplishing specific duties.
Suggested parameters and sets of instructions outlining best practices and standards for accomplishing specific duties.
G
Guidance
Suggested parameters and sets of instructions outlining best practices and standards for accomplishing specific duties.
Suggested parameters and sets of instructions outlining best practices and standards for accomplishing specific duties.
G
Guidance
Suggested parameters and sets of instructions outlining best practices and standards for accomplishing specific duties.
Suggested parameters and sets of instructions outlining best practices and standards for accomplishing specific duties.